
Как
уменьшить грудную клетку
Огромное
количество женщин сталкиваются с
различными дефектами грудных желез,
которые мешают достичь идеальной фигуры.
О том, как устранить те или иные дефекты,
мы расскажем в этой статье.
Немало
женщин имеют от природы широкую грудную
клетку, но сильно заметен этот дефект
только у стройных девушек, особенно
имеющих узкие бедра. И многих из них
интересует вопрос — как
уменьшить грудную клетку.
К сожалению, легкого способа устранить
такой тип грудины не существует. Можно
попытаться немного подтянуть мышцы,
расположенные в этой зоне, однако не
перестарайтесь, ведь при увеличившейся
мышечной массе, спина станет еще шире.
Также часто причина скрывается в лишних
килограммах и отсутствии правильных
пропорций фигуры. Визуально исправить
слишком широкую грудную клетку можно,
убрав лишние жировые отложения и
откорректировав размер грудных желез.
Часто после операции по увеличению
груди грудная клетка уже не выглядит
широкой, а грудь смотрится роскошно.
Непропорциональность
желез встречается довольно часто, но
причин этому может быть несколько.
Бывают случаи, когда одна
грудная мышца больше другой,
и от этого железы выглядят разными по
размеру. В этом случае необходимы
спортивные занятия и укрепление меньшей
по размеру мышцы. В остальных случаях
поможет только помощь пластических
хирургов. Пройти обследование обоих
желез будет не лишним, ведь иногда
причиной увеличения одной из желез
может стать опухоль. Также асимметрия
часто появляется после долгого кормления
грудью, но в основном в этом случае
железы сами восстанавливаются, поэтому
маммопластика в период грудного кормления
запрещена. При помощи пластики можно
как увеличить, так и уменьшить размер
груди, не только сделав их одинаковыми,
но и улучшить их внешний вид, придать
полноту и округлость. Все виды коррекции
груди проходят под общим наркозом, а
период восстановления длится около
месяца, но результат того стоит.
Так как
же увеличить грудные мышцы
девушкам? В случаях, когда
грудь еще не подверглась птозу, несколько
улучшить внешнюю привлекательность
груди можно выполняя специальные
упражнения:
При
постоянных занятиях спортом в течение
продолжительного времени можно как
подтянуть грудные мышцы,
так и немного улучшить форму желез.
Однако быстрых результатов ждать не
стоит, а в более запущенных случаях
отвисания желез достичь заметных
результатов можно только при помощи
хирургической коррекции.
Большая
масса желез, тонкая кожа, отказ от
поддерживающего белья, лишний вес,
возрастные изменения организма все это
способствует птозу, или отвисанию
грудных желез.
Как
исправить птоз молочных желез
Единственно
возможный способ коррекции такого
дефекта – операция. Мастопексия (подтяжка
груди) проводится под общим наркозом,
и занимает от 1 часа, до 4 часов. Во время
коррекции удаляют излишки кожи, тканей
желез, а также подтягивают отвисшие
ткани железы, и фиксируют их к грудной
клетке. В зависимости от степени отвисания
применяют различные разрезы, при
небольшом отвисании – разрез делают
вокруг ареолы и вниз, а когда необходима
более сильная коррекция, применяют
разрезы в форме буквы т, или в виде якоря.
После иссечения всех излишков накладывают
косметические швы. По истечении периода
реабилитации вы сможете снова наслаждаться
подтянутой и высокой грудью. А шрамы со
временем светлеют, и не слишком заметны.
Иметь
красивую, полную грудь – мечта любой
женщины, а как этого достичь зависит от
состояния грудных желез. Некоторым
достаточно правильного питания и
упражнений, другим может помочь только
операция. Если вас беспокоит форма, или
размер молочных желез, то вы можете
прийти на консультацию в клинику и
посоветоваться с врачом о возможных
способах коррекции.
Гипоплазия грудной клетки плода. Хондроэктодермальная дисплазия
При многих скелетных дисплазиях в диспластический процесс вовлекаются ребра и другие кости грудной клетки. Уменьшение размеров грудной клетки приводит к ограничению развития легких и, следовательно, к легочной гипоплазии.
Имеется определенная группа дисплазии, при которых гипоплазия грудной клетки является основным проявлением. К ним относятся асфиксическая дисплазия грудной клетки, синдром Ellis-van Creveld, синдром коротких ребер — полидактилии и кампомелическая дисплазия.
Другими заболеваниями, при которых отмечаются изменения размеров грудной клетки, являются танатофорная дисплазия, ателостеогенез, фиброхондрогенез, ахондрогенез и синдром Jarcho-Levin.
Асфиксическая дисплазия грудной клетки, первоначально описанная М. Jeune et al. и известная как синдром Jeune, является редко встречающимся аутосомно-рецессивным заболеванием. Его распространенность составляет 0,14 на 10 000 родов. Оно характеризуется наличием узкой колоколообразной грудной клетки с короткими, горизонтально расположенными ребрами.
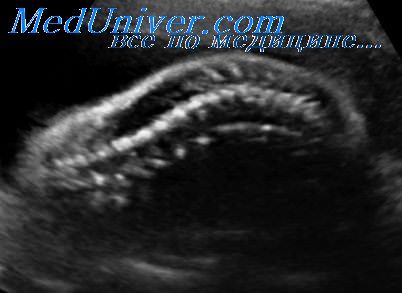
Длинные трубчатые кости бывают нормального размера или слегка укорочены. Характерным признаком, обнаруживаемым при рождении, является наличие оссификации проксимального ядра окостенения бедренной кости, с которым могут сочетаться полидактилия и расщелины губы и/или неба. Дсфиксическая дисплазия грудной клетки имеет широкий спектр клинических проявлений — от летальной до слабо выраженных форм; имеются сообщения о выживших пациентах, страдающих этой патологией.
Клиническое течение заболевания в периоде новорожденности, как правило, осложняется респираторным дистресс-синдромом различной степени тяжести, нефропатией, нарушениями функции печени и поджелудочной железы. Описаны случаи пренатальной диагностики асфиксической дисплазии грудной клетки.
Синдром коротких ребер — полидактилии представляет собой группу заболеваний, характеризующихся микромелией, узкой грудной клеткой и постаксиальной полидактилией. Традиционно различают три различных типа данной патологии (Saldino-Noonan, Majewski и Naumoff). Описаны случаи пренатальной диагностики этих состояний.
Некоторые авторы расширили определение синдрома коротких ребер — полидактилии, чтобы охватить по крайней мере семь состояний, включая три вышеупомянутых, а также заболевания Yang, Marec, Beeraer и асфиксическую дисплазию грудной клетки. J. Spranger и P. Maroteaux считают, что отсутствие полидактилии не исключает наличия данного заболевания.
Хондроэктодермальная дисплазия, также известная как синдром Ellis-van Creveld, наследуется по аутосомно-рецессивному типу. Заболевание характеризуется акромезомелией, нормальным состоянием позвоночника и черепа, постаксиальной полидактилией, удлиненной и узкой грудной клеткой с короткими ребрами и пороками сердца (в 60% случаев). Полидактилия является постоянным признаком заболевания.
Дополнительный палец обычно имеет правильно сформированные пястные и фаланговые кости. Выжившие индивидуумы во взрослом состоянии имеют низкий рост и нормальный интеллект. Описаны случаи пренатальной диагностики хондроэктодермальной дисплазии. Примерно третья часть детей погибает в неонатальном периоде в связи с развитием сердечно-легочной недостаточности.
— Также рекомендуем «Врожденные ампутации у плода. Синдром аглоссии-адактилии»
1. Кампомелическая дисплазия у плода. Диагностика кампомелической дисплазии2. Гипоплазия грудной клетки плода. Хондроэктодермальная дисплазия3. Врожденные ампутации у плода. Синдром аглоссии-адактилии4. Редукционные пороки конечностей у плода. Фокомелия у плода5. Врожденное укорочение бедренной кости. Расщепление кистей и стоп плода6. Аномальная установка кистей плода. Анемия Фанкони у плода7. Синдром TAR у плода. Синдром Aase, Холта-Орама плода8. Ассоциация VATER у плода. Синдром Голденхара и полидактилия плода9. Артрогрипоз у плода. Распространенность артрогрипоза у плода10. Диагностика артрогрипоза плода. Фетальные синдромы
Торакоасфиктическая дистрофия (синдром Жёна)
Торакоасфиктическая дистрофия (синдром Жёна) — редкое генетически гетерогенное заболевание, наследуемое по аутосомно-рецессивному типу, характеризуемое развитием генерализованной остеохондродисплазии, аномалиями костной системы, поражением внутренних органов и глаз. В основе синдрома лежат мутации в генах , расположенных в локусах хромосом 3q25.33, 11q22.3, 4p14, 2q24.3 соответственно, а также мутации в локусе хромосомы 15q13. Эти гены играют важную роль в функционировании цилий в организме млекопитающих. В статье представлены современные данные литературы о генетике, патогенезе, клинической, рентгенологической и КТ-картине, диагностике и терапии этого редкого заболевания. Также впервые в России описана серия наблюдений 7 пациентов с синдромом Жёна. Сочетание таких симптомов, как отставание роста окружности грудной клетки, укорочение конечностей, дыхательная недостаточность, в том числе с кислородозависимостью, рецидивирующие инфекции дыхательных путей, задержка моторного развития, у наблюдавшихся пациентов позволило диагностировать синдром Жёна во всех случаях.
синдром Жёна, торакоасфиктическая дистрофия, генетика, дыхательная недостаточность, кислородозависимость, дети, диагностика, клинические наблюдения
Неонатология: новости, мнения, обучение. 2015. № 4. С. 47-59.
Цилиопатии являются плейотропными заболеваниями и характеризуются такими общими, вариабельными в зависимости от заболевания, клиническими симптомами, как поликистоз почек, печени и поджелудочной железы, дегенерация сетчатки глаза, асимметрия внутренних органов, костные, челюстно-лицевые дефекты и дефекты одонтогенеза, дефекты нервной системы, гидроцефалия, ожирение, бесплодие (табл. 1).
Для патологии костной системы при СЖ характерны низкий рост и деформация грудной клетки, форма которой становится узкой и удлиненной или колоколообразной.
Первые симптомы почечной недостаточности у пациентов проявляются снижением концентрационной функции почек.
Большинство литературных источников указывает на манифестацию поражений внутренних органов у пациентов с СЖ в возрасте старше 2 лет, однако S.N. Reddy и соавт. (2011) описали клиническое наблюдение 3-месячного пациента с диагнозом СЖ с пролонгированным неонатальным холестазом. При объективном исследовании печень и селезенка были увеличены в размерах, твердой консистенции.
При УЗИ брюшной полости выявили гепатоспленомегалию, увеличение общего желчного протока. По данным биопсии печени обнаруживался портальный фиброз с расширением желчных протоков без признаков воспаления или некроза.
T. Hall и соавт. (2009) описали 4-месячного мальчика с СЖ, у которого проявлением патологии желудочно-кишечного тракта стала мальротация кишечника. У ребенка отмечались периодические эпизоды рвоты. Развитие аспирационной пневмонии усугубило тяжелую респираторную патологию.
В табл. 2 обобщены данные о 47 наблюдениях СЖ, представленных в доступной литературе с 1977 по 2012 г.
Дифференциальную диагностику СЖ проводят с ахондроплазией, синдромом Эллиса-ван Кревельда, синдромом коротких ребер — полидактилии типа III и IV, синдромоШвахмана-Даймонда.
Ахондроплазия — системное заболевание скелета, в основе которого лежит замедление роста костей и хрящей.
Помимо хирургических методов лечения пациентам с СЖ проводится симптоматическая терапия. В первую очередь следует обеспечить нормальную дыхательную функцию.
Под нашим наблюдением с 2010 по 2015 г. находилис7 пациентов с этим редким заболеванием, 2 мальчика и 5 девочек, родившихся на 24-40-й неделе гестации (32±5,1) с массой тела при рождении 485-3480 г (1796±1225,9 г), 5 из них были недоношенными (табл. 3). Возраст варьировал от 4 мес до 4 лет, 2 пациентов умерли в возраст11 мес и в 1 год 9 мес. Обследование всех пациентов включало клинический осмотр, антропометрические измерения, стандартные гематологические и, биохимические исследования, рентгенографию органов грудной клетки, ЭхоКГ, УЗИ органов брюшной полости и почек, 2 пациентам была проведена ВРКТ органов грудной клетки. В неонатальном периоде у всех детей наблюдались дыхательные нарушения, требовавшие проведения респираторной терапии. В качестве инициальной респираторной поддержки всем новорожденным проводилась искусственная вентиляция легких (ИВЛ) с переходом на постоянное положительное давление в дыхательных путях через носовые катетеры (nCPAP) в течение 2-33 (16,3±9,2) сут, у 4 пациентов сформировалась тяжелая бронхолегочная дисплазия (БЛД). 4 детей после выписки со II этапа выхаживания нуждались в домашней кислородотерапии, у 3 из них была БЛД. У всех пациентов отмечались частые рецидивирующие респираторные инфекции, ставшие причиной регоспитализаций с развитием тяжелой дыхательной недостаточности (ДН), требовавшей дополнительной оксигенации, 5 детям проводилась повторная ИВЛ.
Клинически у всех пациентов были обнаружены отставание роста окружности грудной клетки, неравномерное укорочение конечностей (рис. 1), у 2 детей печень и селезенка пальпировались на 3-4 см ниже края реберной дуги. По данным антропометрии, рост всех пациентов был ниже 50-го центиля. Окружность головы при рождении у наблюдавшихся пациентов составляла 33, 22, 32, 28, 22,5, 24, 34 см, окружность грудной клетки 31, 19, 30, 22, 17, 18, 29 см соответственно. У 5 пациентов окружность головы в возрасте 9 мес составляла 46, 36, 45, 40, 35 см, грудной клетки — 41, 33, 42, 37, 33 см соответственно. 2 детей из нашей серии наблюдений не достигли возраста 10 мес, окружность головы в возрасте 6 и 4 мес у них составила 33 и 40 см, грудной клетки — 29 и 33 см соответственно. Известно, что в норме в возрасте 3 мес эти показатели сравниваются и в течение всей жизни преобладают показатели окружности грудной клетки. У наблюдавшихся пациентов с СЖ в любом возрасте окружность головы преобладала над окружностью грудной клетки. У всех детей отмечались гипотрофия II-III степени и задержка темпов психомоторного развития.
На рентгенограммах органов грудной клетки у всех пациентов были выявлены типичные признаки СЖ: узкая грудная клетка, горизонтально расположенные ребра, высокая конфигурация ключиц, имеющих вид велосипедного руля. У 1 пациента обнаружены деформация костей таза и дисплазия обоих тазобедренных суставов.
В серии наблюдений обращает на себя внимание большое число недоношенных детей, страдающих БЛД (4 и7 пациентов), которая у 3 детей осложнилась высокой ЛГ.
Представляем наблюдение доношенного ребенка с СЖ, которому была выполнена ВРКТ грудной клетки (пациент 3). Новорожденная девочка С. после рождения переведена в отделение реанимации и интенсивной терапии (ОРИТ) на 1-е сутки жизни с направляющим диагнозом: внутриутробная пневмония, множественные пороки развития.
Из анамнеза известно, что ребенок от соматически здоровой женщины 32 лет, от 2-й беременности (1-я — самопроизвольные роды на 38-й неделе гестации, мальчик, 5 лет, здоров), протекавшей с угрозой прерывания во II триместре. Роды самопроизвольные, срочные, на 40-й неделе гестации, масса тела при рождении 3200 г, рост 51 см, окружность голов32 см, груди — 30 см, оценка по шкале Апгар 6/7 баллов.
Состояние при рождении расценено как тяжелое, обусловленное ДН, течением внутриутробной пневмонии. Девочка в 1-е сутки жизни была интубирована, переведена на ИВЛ, экстубирована на 3-и сутки жизни. На 4-е сутки жизни ребенок переведен на II этап выхаживания.
Девочка неоднократно, каждые 1-2 мес, находилась на стационарном лечении в разных больницах Москвы, причиной госпитализации стали рецидивирующие респираторные инфекции, в том числе потребовавшие проведения кислородотерапии (в возрасте 8 мес — правосторонняя пневмония, в 9 мес — острый бронхит, в 1 год 2 мес — двусторонняя пневмония, в 1 год 4 мес — двусторонняя полисегментарная пневмония, в 1 год 5 мес — острый бронхит).
В возрасте 6 мес осмотрена генетиком, по фенотипическому статусу заподозрен СЖ, генетическое исследование не проводилось.
В возрасте 1 года 8 мес была госпитализирована в ОРИТ, где при обследовании поставлен диагноз «двусторонняя полисегментарная пневмония». ДН III степени. Кислородозависимость. Синдром Жёна, формирующийся пневмосклероз.
Белково-энергетическая недостаточность III степени. Кахексия. На момент поступления при осмотре: тургор тканей резко снижен, грудная клетка резко уменьшена, деформирована, конечности укорочены, видимые слизистые оболочки сухие, цианотичные. Масса тела 4800 г, рост 72 см, окружность головы — 46 см, окружность груди 43 см. ЧДД 50 в минуту. SatO62%. Дыхание самостоятельное с резким втяжением уступчивых мест грудной клетки, отмечалось раздувание крыльев носа. Аускультативно дыхание жесткое, проводится с обеих сторон, ослаблено в нижних отделах слева, с двух сторон выслушиваются разнокалиберные хрипы. При санации из носоглотки аспирируется небольшое количество слизистой мокроты. ЧСС 164 в минуту.
АД 80/40 мм рт.ст. Тоны сердца приглушены, ритмичные, систолический шум на верхушке. Границы относительной сердечной тупости не расширены. Живот не вздут, доступен глубокой пальпации. Печень увеличена, пальпируется на 5 см ниже края реберной дуги, селезенка — на 1 см. По данным рентгенографии органов грудной клетки визуализировались изменения, типичные для СЖ — колоколообразная деформация грудной клетки, горизонтальное расположение ребер, ключицы по типу велосипедного руля (рис. 2). При проведении ВРКТ определялись признаки уменьшения объема легких, двусторонней полисегментарной пневмонии на фоне двусторонних рубцово-фиброзных изменений, формирующегося пневмосклероза (рис. 3). За время пребывания в ОРИТ девочка находилась на ИВЛ в течении 5 сут.
В возрасте 1 года 9 мес у ребенка резко наросли симптомы ДН, в связи с чем девочка вновь была госпитализирована в ОРИТ. При поступлении отмечалось спонтанное, неадекватное, поверхностное, аритмичное дыхание. Одышка смешанного характера с преобладанием инспираторного компонента, с участием в акте дыхания вспомогательной мускулатуры и втяжением уступчивых мест грудной клетки, также отмечалось раздувание крыльев носа. Оксигенация по данным пульсоксиметра неудовлетворительная. Девочка была интубирована, проводилась ИВЛ. При обследовании поставлен диагноз: CЖ. РСВ-инфекция, острый бронхиолит, тяжелое течение. Двусторонняя полисегментарная пневмония. ДН III степени. Белково-энергетическая недостаточность III степени. Кахексия. Анемия. Отмечалась отрицательная клиническая динамика: ухудшение состояния, сатурация снижалась на 10-20% каждые 4 ч, достигнув значения 25-0%. АД не определялось. Проводимые реанимационные мероприятия были неэффективными, зафиксирована биологическая смерть.
В представленном наблюдении комбинация таких диагностических критериев, как отставание показателей роста и окружности грудной клетки от нормальных величин, укорочение конечностей, ДН, рецидивирующие респираторные инфекции, задержка психомоторного развития, позволила установить у ребенка диагноз «синдром Жёна». Данный диагноз необходимо предполагать у дисморфичных пациентов, нуждающихся в проведении оксигенотерапии в неои постнеонатальном периодах, включая домашнюю оксигенотерапию. Очень важно исключать данное заболевание у кислородозависимых детей, поскольку СЖ может быть одной из причин как острой, так и пролонгированной кислородозависимости.
1. Jeune M., Beraud C., Carron R. Dystrophie thoracique asphyxiante de caractere familial // Arch. Fr. Pediatr. 1955. Vol. 12. P. 886-891.
2. Phillips J.D., Van Aalst J.A. Jeune’s syndrome (asphyxiating thoracic dystrophy): congenital and acquired // Semin. Pediatr. Surg. 2008. Vol. 17, N 3. P. 167-172.
3. Morgan N.V., Bacchelli C., Gissen P. et al. A locus for asphyxiating thoracic dystrophy, ATD maps to chromosome 15q13 // J. Med. Genet. 2003. Vol. 40. P. 431-435.
4. Beales P.L., Bland E., Tobin J.L. et al. IFT80, which encodes a conserved intraflagellar transport protein, is mutated in Jeune asphyxiating thoracic dystrophy // Nat. Genet. 2007. Vol. 39. P. 727-729.
5. Dagoneau N., Goulet M., Genevieve D. et al. DYNC2H1 mutations cause asphyxiating thoracic dystrophy and short rib-polydactyly syndrome, type III // Am. J. Hum. Genet. 2009. Vol. 84. P. 706-71.
6. Jones K.L. Smith’s Recognizable Patterns of Human Malformation. 6th ed. Philadelphia, PA : Elsevier; Saunders, 2006. P. 450-452.
7. Badano J.L., Mitsuma N., Beale P. et al. The ciliopathies: an emerging class of human genetic disorders // Annu. Rev. Genomic Hum. Genet. 2006. Vol. 7. P. 125-148.
8. Satir P., Pedersen L.B., Christensen S.T. The primary cilium at a glance // J. Cell Sci. 2010. Vol. 123. P. 499-503.
9. Ferkol T. Primary ciliary dyskinesia (Immotile cilia syndrome) // Nelson Textbook of Pediatrics. 19th ed. / eds R.M. Kliegman, B.F. Stanton, J.W. St. Geme III, N.F. Schor et al. Philadelphia, PA : Elsevier; Saunders, 2011. P. 1497e2-1497e6.
10. Huber C., Cormier-Daire V. Ciliary disorder of the skeleton // Am. J. Med. Genet. Pt C (Seminars in Medical Genetics). 2012. Vol. 160C.
11. McInerney-Leo A.M., Schmidts M., Cortes C.R. et al. Short-rib polydactyly and Jeune syndromes are caused by mutations in WDR60 // Am. J. Hum. Genet. 2013. Vol. 93, N 3. P. 515-523.
12. Schmidts M., Vodopiutz J., Christou-Savina S. et al. Mutations in the gene encoding IFT dynein complex component WDR34 cause Jeune asphyxiating thoracic dystrophy // Am. J. Hum. Genet. 2013. Vol. 93, N 5.
13. Baujat G., Huber C., Hokayem E.J. et al. Asphyxiating thoracic dysplasia: clinical and molecular review of 39 families // J. Med. Genet. 2013. Vol. 50. P. 91-98.
14. Barnes N.D., Hull D., Simons J.S. Thoracic dystrophy // Arch. Dis. Child. 1969. Vol. 44. P. 11-17.
15. Cortina H., Beltran J., Olague R. et al. The wide spectrum of the asphyxiating thoracic dysplasia // Pediatr. Radiol. 1979. Vol. 8. P. 93-99.
16. Friedman J.M., Kaplan H.G., Hall J.G. The Jeune syndrome (asphyxiating thoracic dystrophy) in an adult // Am. J. Med. 1975. Vol. 59.
17. Hennekam R.C.M., Beemer F.M., Gerards L.J., Cats B. Thoracic pelvic phalangeal dystrophy (Jeune syndrome) // Tijdschr. Kindergeneeskd. 1983. Vol. 51. P. 95-100.
18. Herdman R.C., Langer L.O. The thoracic asphyxiant dystrophy and renal disease // Am. J. Dis. Child. 1968. Vol. 116. P. 192-201.
19. Kajantie E., Andersson S., Kaitila I. Familial asphyxiating thoracic dysplasia: clinical variability and impact of improved neonatal intensive care // J. Pediatr. 2001. Vol. 139. P. 130-133.
21. Razzi A., Rosso C., Durand P. Anatomopathological contribution to asphyxiating thoracic dystrophy of the unweaned child (Jeune’s disease) find considerations on the therapeutic usefulness of a surgical operation on the ribs // Panminerva Med. 1965. Vol. 8. P. 444-449.
22. Tuysuz B., Baris S., Aksoy F., Madazh R. et al. Clinical variability of asphyxiating thoracic dystrophy (Jeune) syndrome: evaluation and classification of 13 patients // Am. J. Med. Genet A. 2009. Vol. 149, N 8. P. 1727-1733.
23. Langer L.O. Thoracic-pelvic-phalangeal dystrophy: Asphyxiating thoracic dystrophy of the newborn, infantile thoracic dystrophy // Radiology. 1968. Vol. 91. P. 447-456.
24. Ochs M., O’Brodovich H. The structural and physiologic basis for respiratory disease // Kendig and Chernick’s Disorders of the RespiratorTract in Children. 8ed. / eds R.W. Wilmott, T.F. Boat, A. Bush, V. Chernick et al. Philadelphia, PA : Elsevier; Saunders, 2012. P. 35-74.
25. Boas S.R. Skeletal diseases influencing pulmonary function // Nelson Textbook of Pediatrics. 19th ed. / eds R.M. Kliegman, B.F. Stanton, J.W. St. Geme III, N.F. Schor et al. Philadelphia, PA : Elsevier; Saunders. 2011. P. 1516-1518.
26. Keppler-Noreuil K.M., Adam M.P., Welch J., Muilenburg A., WillinM.C. Clinical insights gained from eight new cases and review of reported cases with Jeune syndrome (asphyxiating thoracic dystrophy) // Am. J. Med. Genet A. 2011. Vol. 155A, N 5. P. 1021-1032.
27. Chen H. Medscape Reference. Genetics of asphyxiating thoracic dystrophy (Jeune syndrome). URL: http://emedicine.medscape.com/article/945537 — overview. Updated June 27, 2013. Accessed July 22, 2013.
28. Paladini D., Volpe P. Ultrasound of Congenital Fetal Anomalies: Differential Diagnosis and Prognostic Indicators. Informa Medical, 2007.
29. Lehman A.M., Eydoux P., Doherty D., et al. Co-occurence of Joubert syndrome and Jeune asphyxiating thoracic dystrophy // Am. J. Hum. Gene A. 2010. Vol. 152A, N 6. P. 1411-1419.
30. Donaldson M.D.C., Warner A.A., Trompeter R.S. et al. Familial juvenile nephronophtisis, Jeune’s syndrome, and associated disorders // Arch. Dis. Child. 1985. Vol. 60. P. 426-434.
31. Edelson P.J., Spackman T.J., Belliveau R.E., Mahoney M.J. A renal lesion in asphyxiating thoracic dysplasia // Birth Defects Orig. Artic Ser. 1974. Vol. 10, N 4. P. 51-56.
32. Giorgi P.L., Gabrielli O., Bonifazi V. et al. Mild form of Jeune syndrome in two sisters // Am. J. Med. Genet. 1990. Vol. 35. P. 280-282.
33. Gruskin A.B., Baluarte H.J., Cote M.L., Elfenbein I.B. The renal disease of thoracic asphyxiant dystrophy // Birth Defects Orig. Artic Ser. 1974. Vol. 10, N 4. P. 44-50.
34. Ozcay F., Derbent M., Demirhan B. et al. A family with Jeune syndrom// Pediatr. Nephrol. 2001. Vol. 16. P. 623-626.
35. Shah K.J. Renal lesion in Jeune’s syndrome // Br. J. Radiol. 1980. Vol. 53. P. 432-436.
36. Lacson A., Bernstein J., Risdon R.A., Gilbert-Barness E. Renal system // Potter’s Pathology of the Fetus, Infant, and Child. 2nd ed. / ed. E. Gilbert-Barness. Philadelphia, PA : Mosby; Elsevier, 2007. P. 1281-1373.
38. Russo P. Liver including tumors, gallbladder, and biliary tree // Potter’s Pathology of the Fetus, Infant, and Child. 2nd ed. / ed. E. GilbertBarness. Philadelphia, PA : Mosby; Elsevier, 2007. P. 1207-1280.
39. Reddy S.N., Seth B.A., Colaco P. Jeune syndrome with neonatal cholestasis // Indian J. Pediatr. 2011. Vol. 78, N 9. P. 1151-1153.
40. Keogh S., McKee S., Smithson S., Grier D., Steward C. ShwachmanDiamond syndrome: a complex case demonstrating the potential for misdiagnosis as asphyxiating thoracic dystrophy (Jeune syndrome) // BMPediatr. 2012. Vol. 12. P. 48.
41. Casteels I., Demandt E., Legius E., Visual loss as the presenting sign of Jeune syndrome // Eur. J. Paediatr. Neurol. 2000. Vol. 4. P. 243-247.
42. Hall T., Bush A., Fell J., Offiah A. et al. Ciliopathy spectrum expanded. Jeune syndrome associated with foregut dysmotility and malrotatio// Pediatr. Pulmonol. 2009. Vol. 44, N 2. P. 198-201.
43. Suita S., Taguchi T., Ieiri S., Nakatsuji T. Hirschsprung’s disease in Japan: analysis of 3852 patients based on a nationwide survey in 30 years // J. Pediatr. Surg. 2005. Vol. 40. P. 197-202.
44. Rahmani R., Sterling C.L., Bedford H.M. Prenatal ultrasound of Jeune-like syndromes with two-dimensional and three-dimensional sonograph// J. Clin. Ultrasound. 2011. Vol. 40, N 4. P. 222-226.
45. Chen C.P., Lin S.P., Liu F.F. et al. Prenatal diagnosis of asphyxiating thoracic dysplasia (Jeune syndrome) // Am. J. Perinatol. 1996. Vol. 13.
47. Zimmer E.Z., Weinraub Z., Raijman A. et al. Antenatal diagnosis of a fetus with an extremely narrow thorax and short limb dwarfism // Clin. J. Ultrasound. 1984. Vol. 12. P. 112-114.
48. Tonni G., Panteghini M., Bonasoni M.P., Pattacini P. et al. Prenatal ultrasound and MRI diagnosis of Jeune syndrome Type I (asphyxiating thoracic dystrophy) with histology and post-mortem three-dimensional CT confirmation // Fetal Pediatr. Pathol. 2013. Vol. 32, N 2. P. 123-132.
49. URL://www.orpha.net/consor/cgibin/ClinicalLabs_Search.php?lng=EN&type_list=clinicalLabs_search_simple_shd&data_id=283&Disease(s)/group%20of%20diseases=Jeune-syndrome&search=ClinicalLabs_Search_Simple&ChdId=283&ClinicalLabs_ClinicalLabs_Search_di seaseGroup=Jeunesyndrome&ClinicalLabs_ClinicalLabs_Search_diseaseType=Pat&ClinicalLabs_ClinicalLabs_Search_ country=NN&ClinicalLabs_ClinicalLabs_Search_GeographicType=null&C linicalLabs_ClinicalLabs_Search_ClinicalLabsList_Sort=QoS (
50. Dror Y., Freedman M.H. Shwachman-diamond syndrome // Br. J. Haematol. 2002. Vol. 118. P. 701-713.
51. Oberklaid R., Danks D.M., Mayne V., Campbell P. Asphyxiating thoracic dysplasia. Clinical, radiological, and pathological information on 10 patients // Arch. Dis. Child . 1977. Vol. 52. P. 758-765.
52. Defendi G.L. Medscape Reference. Genetics of achondroplasia: background. URL: http://emedicine.medscape.com/article/941280 — overview. Updated April 16, 2012. Accessed July 22, 2013.
53. Afsharpaiman S., Saburi A., Waters K.A. Respiratory difficulties and breathing disorders in achondroplasia // Paediatr. Respir. Rev. 2013. Vol. 14, N 4. P. 250-255.
54. Rosenberg A.E., Nielsen G.P., Krishnasetty V., Rosenthal D. Disorders of the skeletal system including tumors // Potter’s Pathology of thFetus, Infant, and Child. 2nd ed. / ed. E. Gilbert-Barness. Philadelphia, PA : Mosby; Elsevier, 2007. P. 1797-1897.
55. Dror Y., Freedman M.H.: Shwachman-Diamond syndrome: an inherited preleukemic bone marrow failure disorder with aberrant hematopoietic progenitors and faulty marrow microenvironment // Blood. 1999. Vol. 94.
56. Waldhausen J.H.T., Redding G.J., Song K.M. Vertical expandable prothetic titanium rib for thoracic insufficiency syndrome: a new method to treat an old problem // J. Pediatr. Surg. 2007. Vol. 42, N 1. P. 76-80.
57. Conroy E., Eustace N., Mccormack D., Sternoplasty and rib distraction in neonatal Jeune syndrome // J. Pediatr. Orthop. 2010. Vol. 30, N 6.
58. Kikuchi N., Kashiwa H., Toshihoko O., Kato M. et al. The Nuss technique for Jeune asphyxiating thoracic dystrophy repair in siblings // Ann. Plast. Surg. 2010. Vol. 65, N 2. P. 214-218.
59. Davis J.T., Heistein J.B., Castile R.G. et al. Lateral thoracic expansion for Jeune syndrome: Midterm results // Ann. Thorac. Surg. 2001. Vol. 72, N 3. P. 872-877.
60. Davis J.T., Long F.R., Adler B.H., Castile R.G. et al. Lateral thoracic expansion for Jeune syndrome: evidence of rib healing and new bone formatio// Ann. Thorac. Surg. 2004. Vol. 77, N 2. P. 445-448.
61. Davis J.T., Ruberg R.L., Lippink D.M., McCoy K.S. et al. Lateral thoracic expansion for Jeune’s asphyxiating dystrophy: a new approach // Ann. Thorac. Surg. 1995. Vol. 60, N 3. P. 694-696.
62. Chen H. Genetic Diagnosis and Counseling. 2nd ed. N.Y. : Springer, 2012. P. 157-166.
63. Labrune P., Fabre M., Trioche P., Estournet-Mathiaud B. et al. Jeune syndrome and liver disease: report of three cases treated with ursodeoxycholic acid // Am. J. Med. Genet. 1999. Vol. 87. P. 324-328.
64. Дегтярева Е.А., Овсянников Д.Ю., Зайцева Н.О., Шокин А.А. Легочная гипертензия и легочное сердце у детей с бронхолегочной дисплазией: факторы риска, диагностика, возможности терапии и профилактики // Педиатрия. . 92, № 5.
65. Drera B., Ferrari D., Cavalli P., Poggiani C. A case of neonatal Jeune syndrome expanding the phenotype // Clin. Case Rep. 2014. Vol. 2, N 4. P. 156-158.